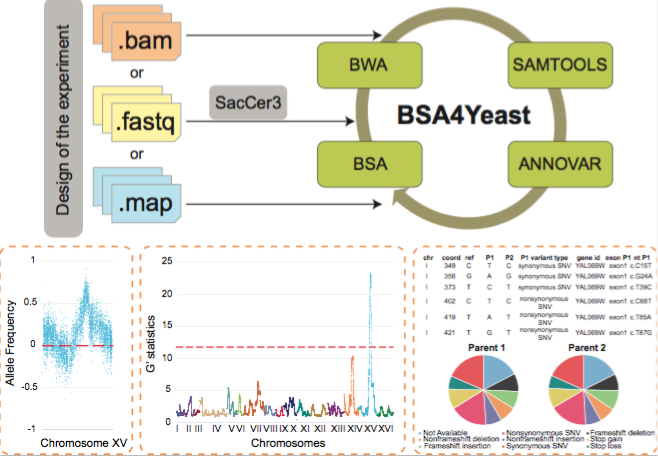
About BSA4Yeast
BSA4Yeast is a comprehensive web application that performs QTL
(Quantitative Trait Loci) mapping using bulk segregant analysis
(BSA) of yeast sequencing data. It has the following main
advantages and features:
A step-by-step description of the software is provided in the Use tab to facilitate QTL analyses.
Yeast (S. cerevisiae) genomics
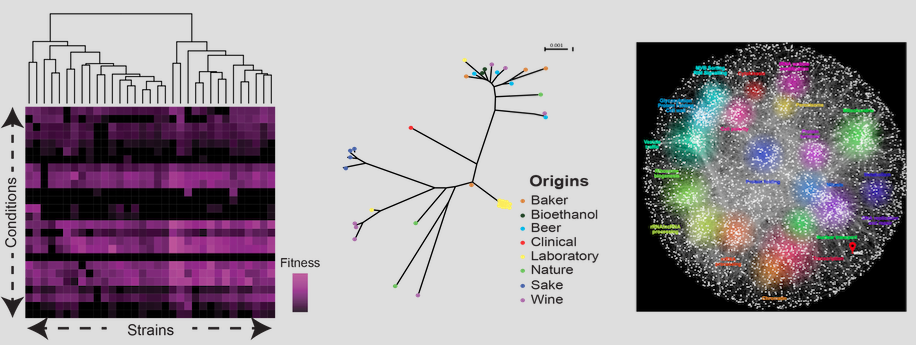
The baker's yeast (Saccharomyces cerevisiae) has been used since decades in research studies on genetics, cell biology and more recently genomics. It is a model organism for population genomics than can help to gain a better understanding of genotype-phenotype relationships. Trait profiling and genome analyses show a broad phenotypic and genetic variability in this organism. Thus, it is suitable for deciphering the genetic architecture of simple and complex traits.
QTL analysis (Quantitative Trait Locus analysis)
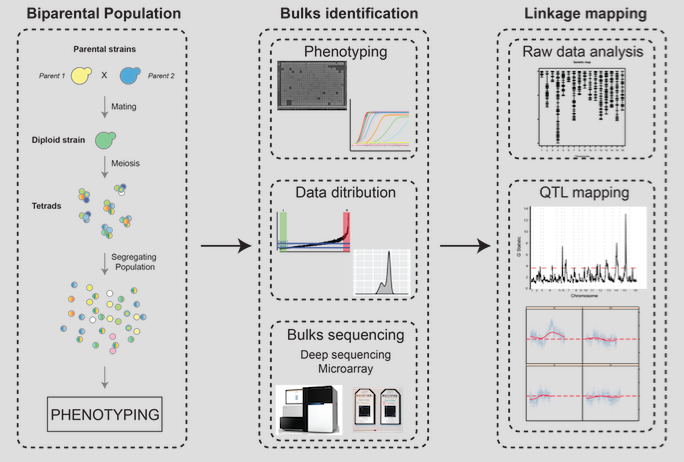
Linkage mapping is a statistical approach to decipher the genetic architecture of simple and complex traits. It relies on the investigation of a progeny obtained by crossing two distinct individuals. In Bulk Segregant Analysis (BSA) based QTL mapping, deep sequencing genotyping, using next generation sequencing technologies, and mapping against genetic markers determined for the parental individuals enables the identification of genes associated with a phenotype of interest. One- or two-tailed bulk design can be applied: A one-tailed bulk design compares of samples from one extreme phenotype (e.g. short lifespan) vs. the entire distribution of phenotypes; a two-tailed bulk design compares samples from two extreme phenotypes against each other (e.g. short lifespan vs. long lifespan).
NGS (Next Generation Sequencing)
NGS (Next Generation Sequencing) is a new nucleic acid sequencing approaches emerged allowing to obtain fast and reliable data in a cost-efficient manner. Since 2000, the compact yeast genomes (~12Mb) serve as milestone for comparative genomics and recently for investigating intraspecies variation and the genotype-phenotype relationship combining both NGS and powerful statistical methods.
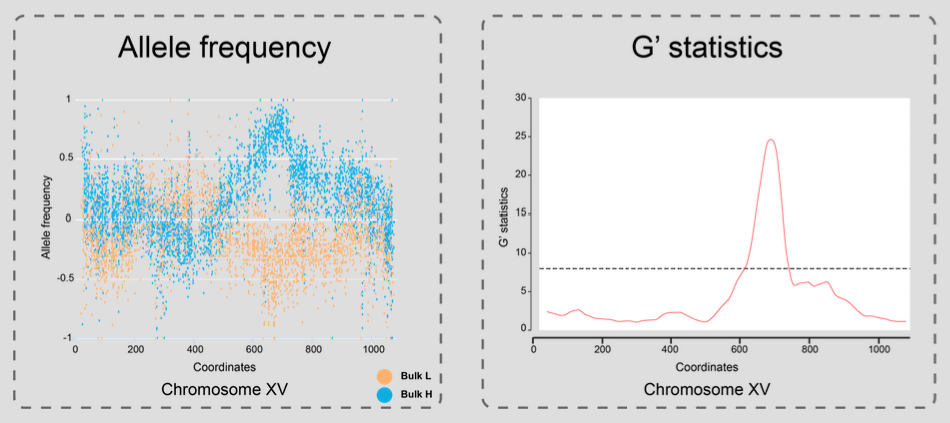
G' statistics