1) Please specify your input data file for uploading.
2) Please wait until your input file has been uploaded.
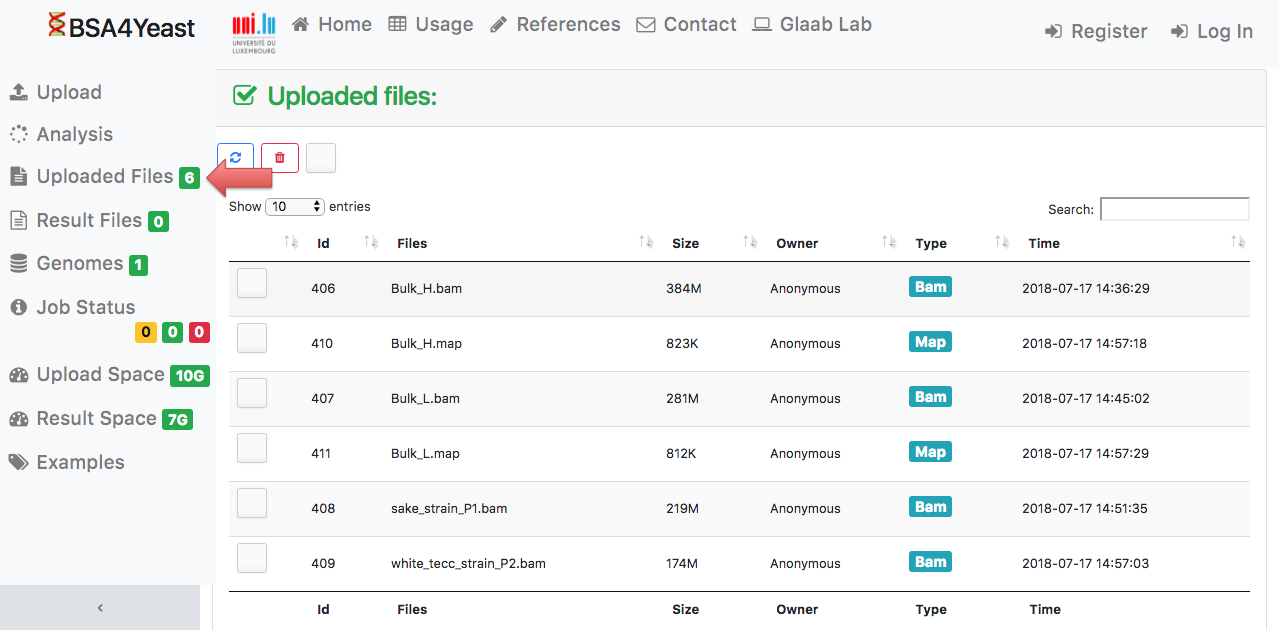
3) Check the uploaded files.
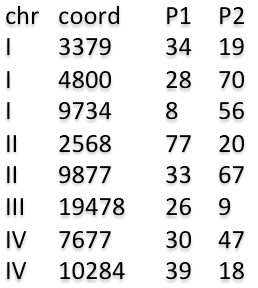
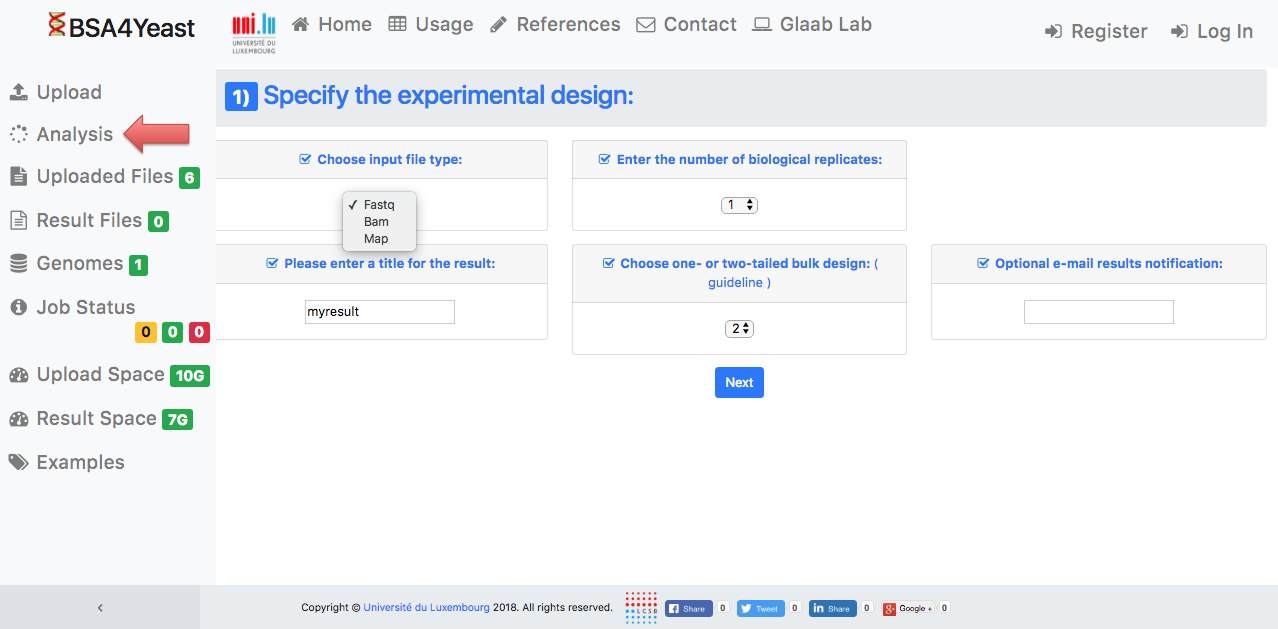
4) Set up your experimental design.
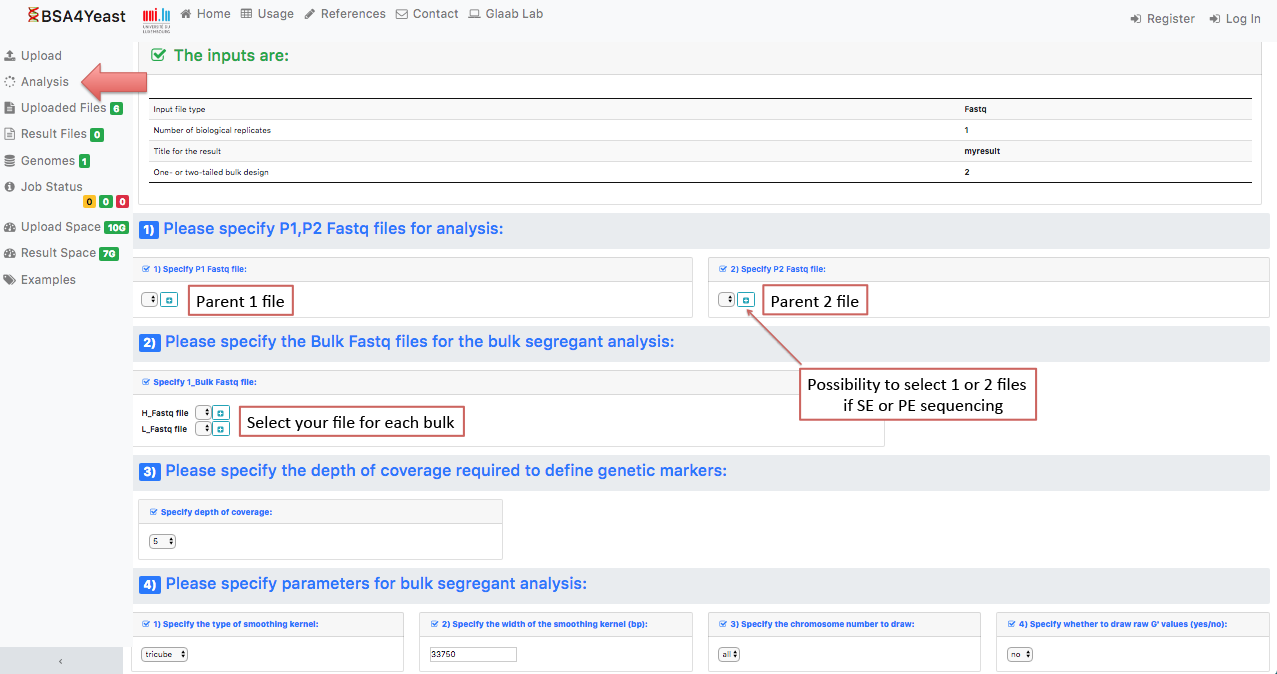
5.1) Select and define parameters for .fastq files.
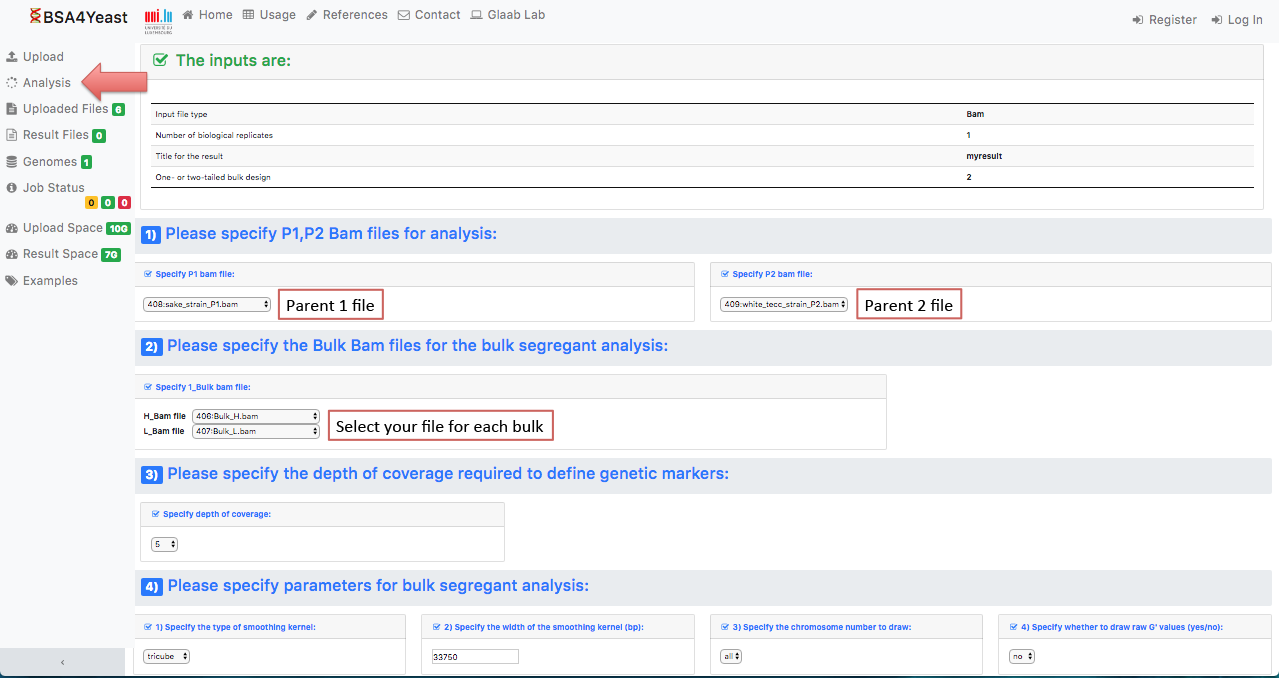
5.2) Select and define parameters for .bam files.
5.3) Select and define parameters for .map files.
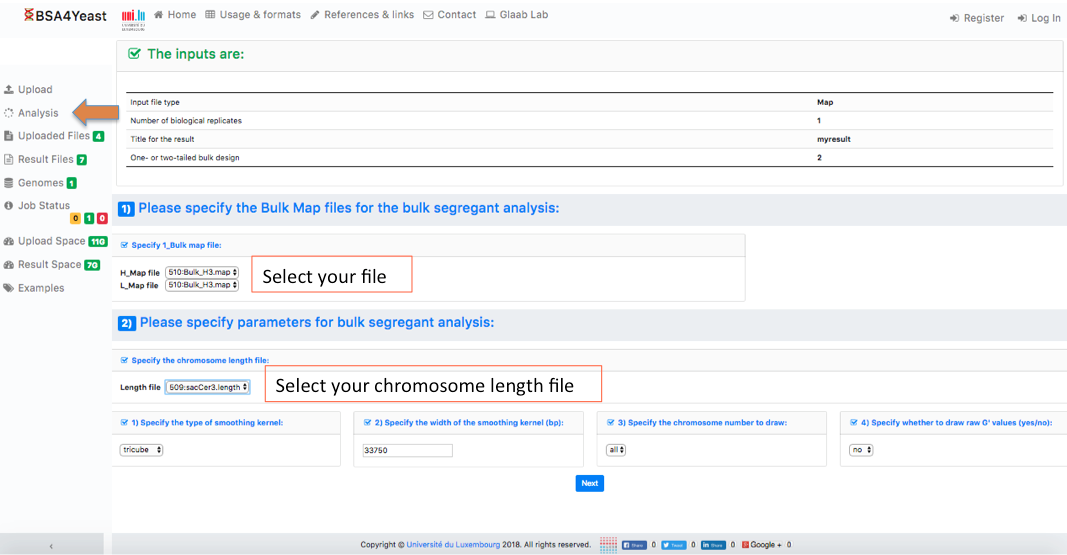
6) Check your settings and click on Calculate G' button.

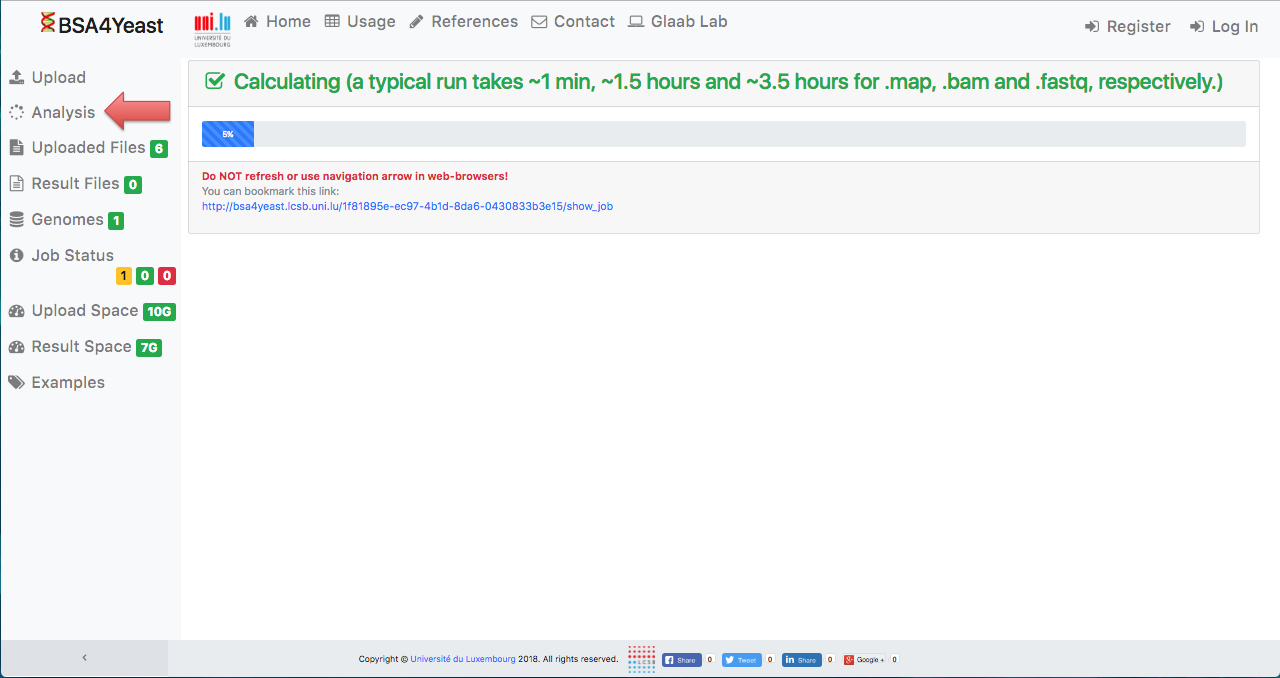
7) Check progress.
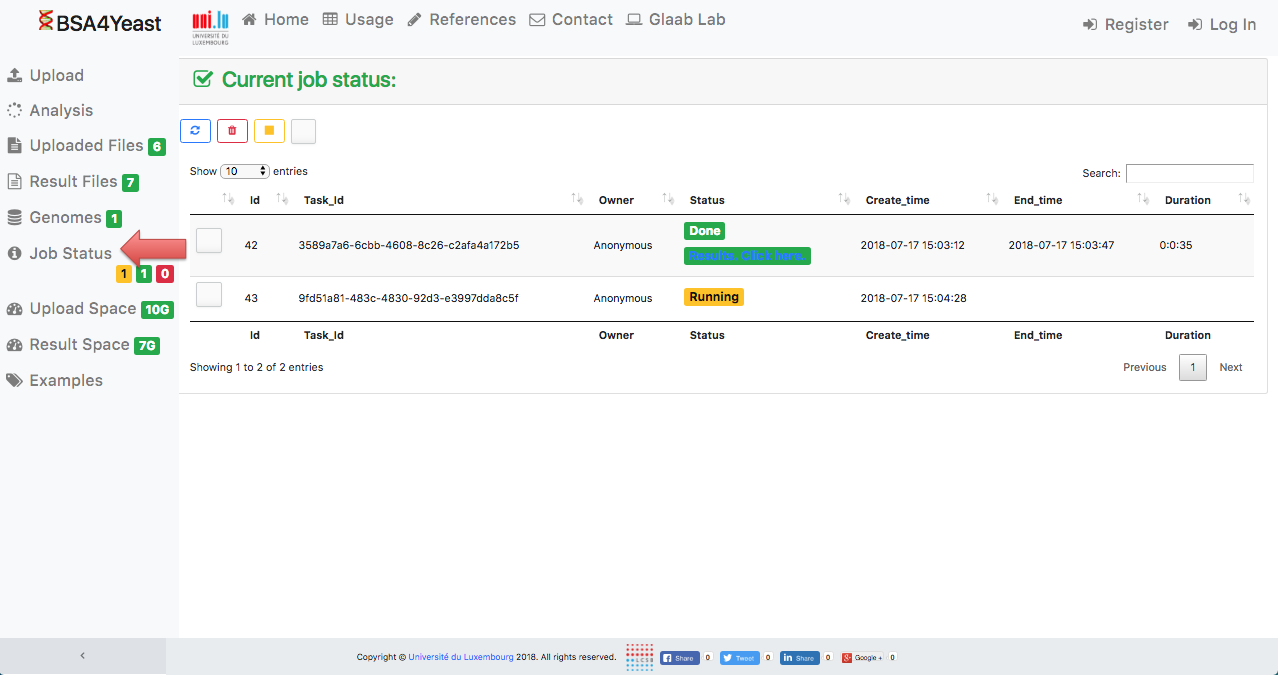
8) Check job status.
9) Optionally receive an e-mail notification when the analysis is complete.
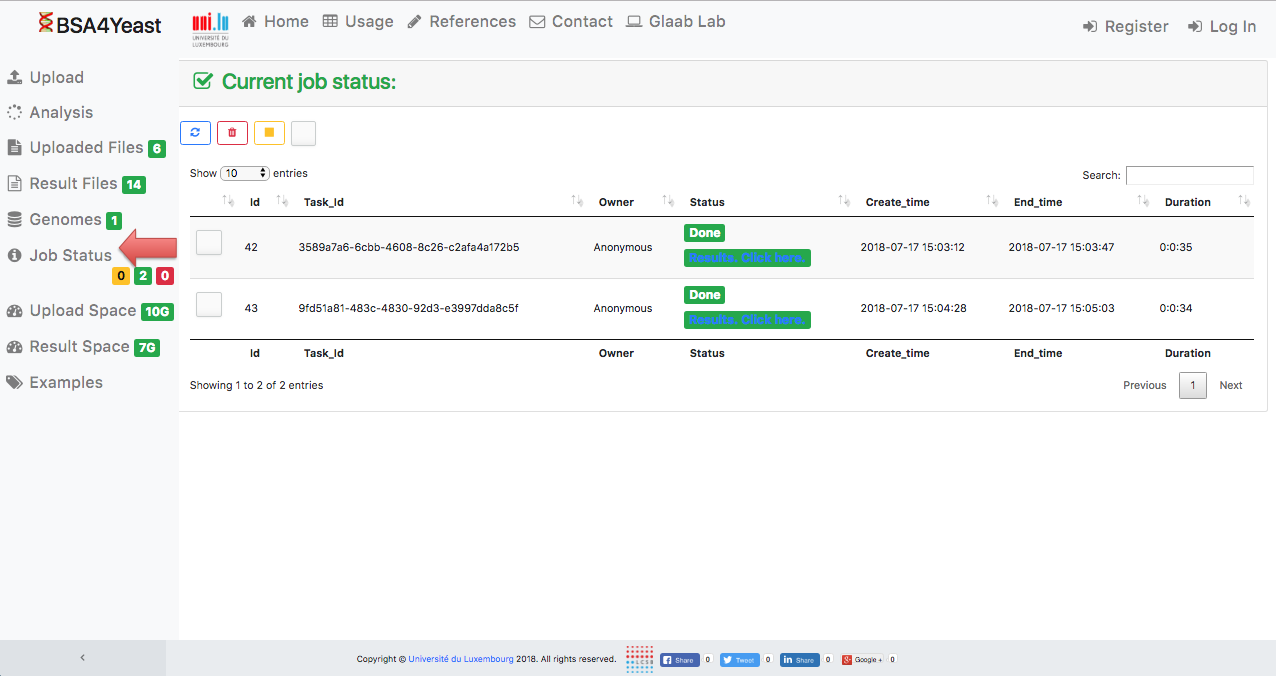
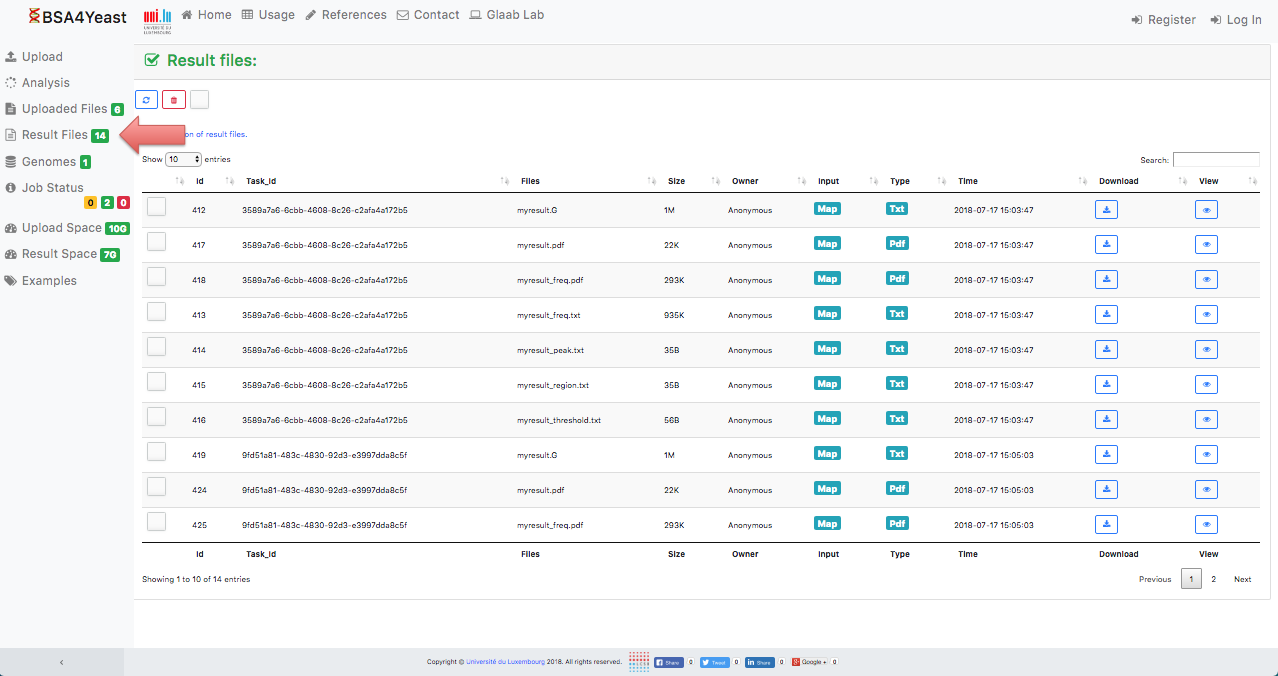
10) Result files can be downloaded.
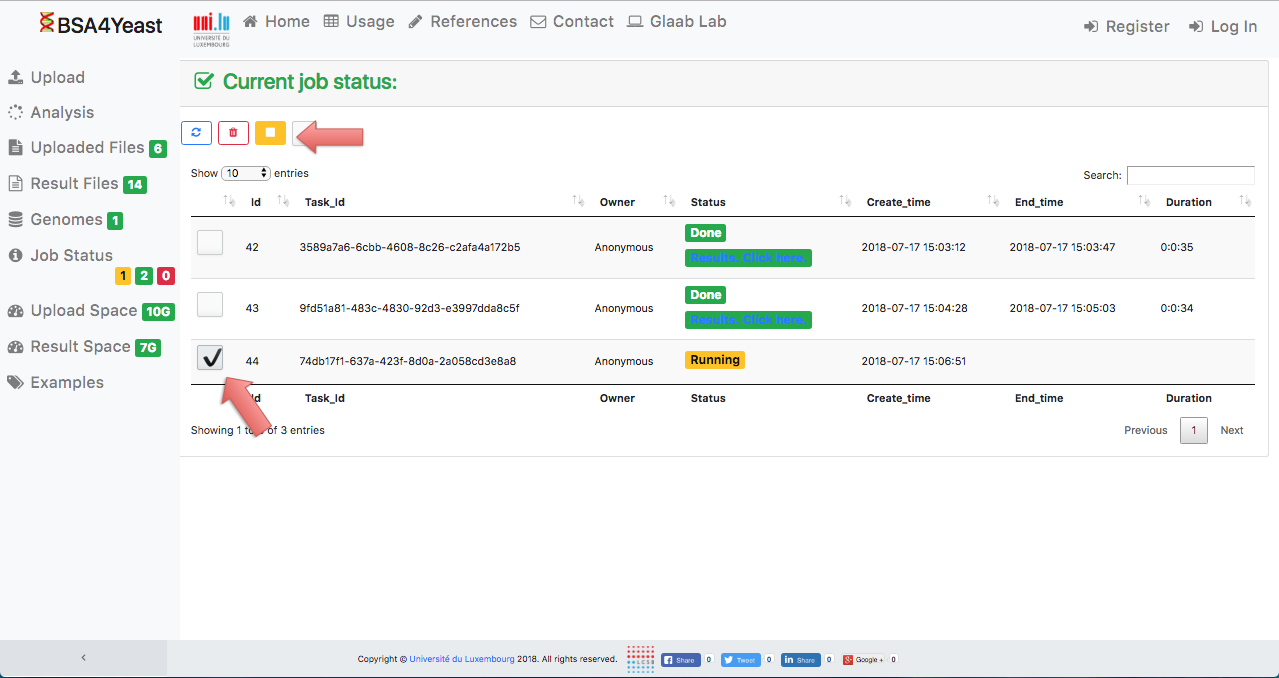
11) Each analysis can be manually stopped.
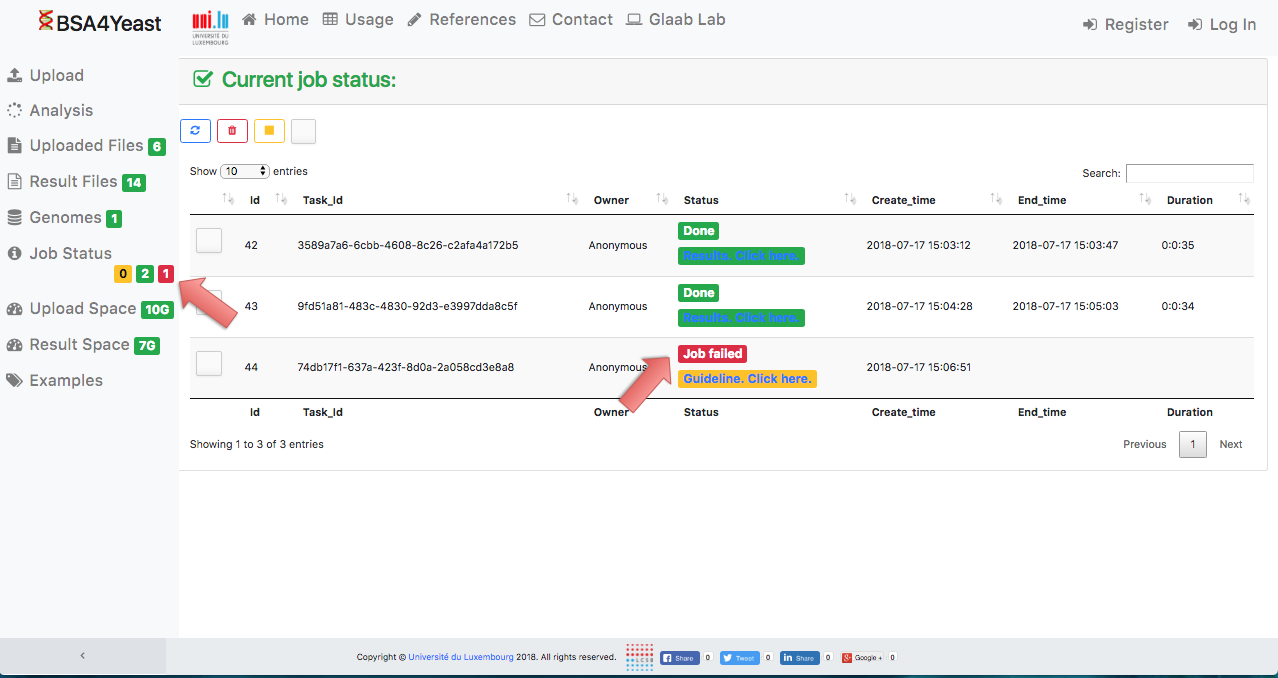
12) Stopped analyses will be displayed as "Failed".
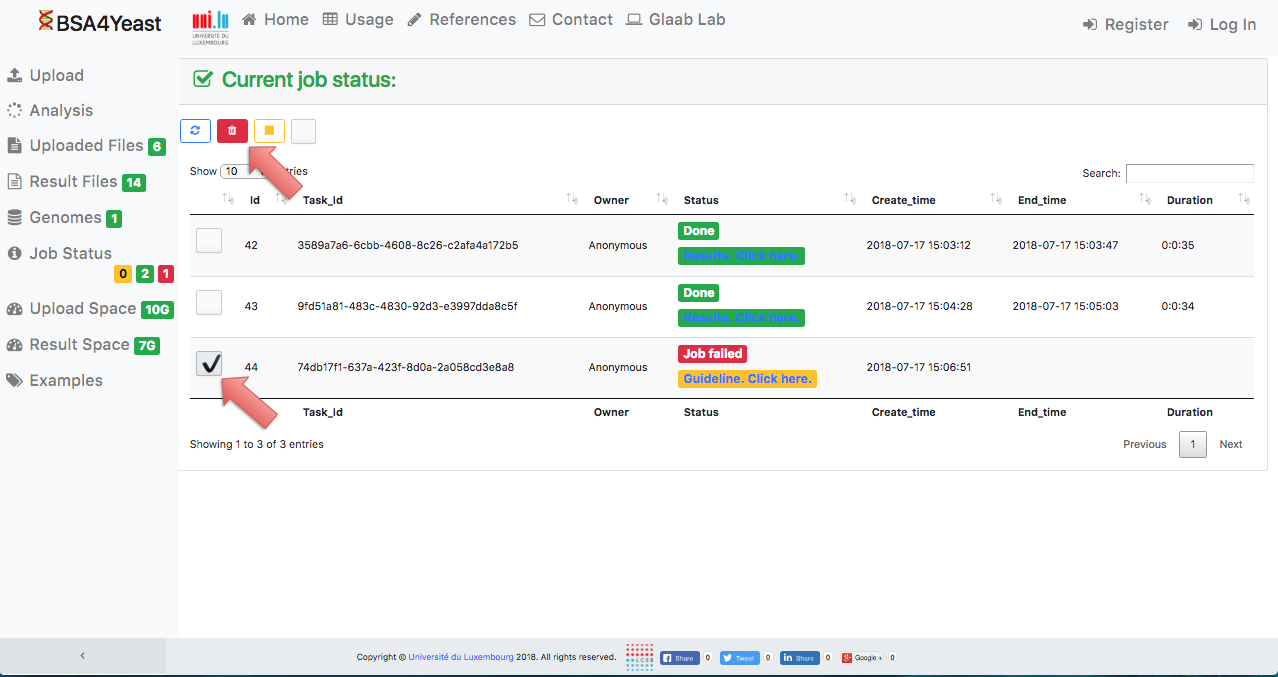
13) Each analysis can be manually deleted.
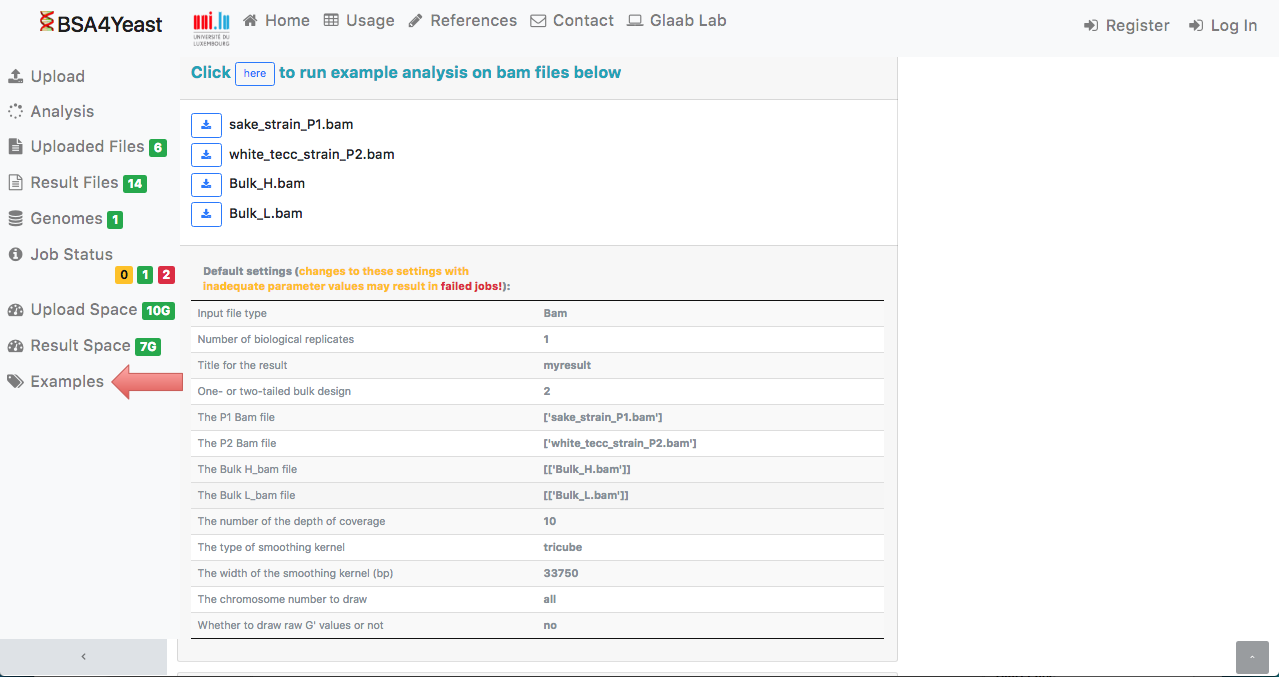
14) In the webpage, by clicking on the "Example" tab,
the user can either run an example dataset
with default settings or download an example dataset and run the analysis with user-defined settings.